
tmap: An Integrative Framework Based On Topological Data Analysis For Population
Date:06-01-2020 | 【Print】 【close】
With the advent of next generation sequencing technologies, conventional data analysis methods are becoming inadequate for the analysis of highly complex and voluminous microbiome data. In particular, nonlinear associations hidden in a high-dimensional microbiome dataset are hard to capture using common linear-based methods. Furthermore, current population-scale microbiome studies usually deal with complex variations of microbiome associated with large-scale host phenotypes or environment types, such as the American Gut Project and the Earth Microbiome Project. To enable the discovery of underappreciated but significant host-associated patterns from comprehensive microbiome sequencing and sample phenotype metadata, it is urgent to develop novel and effective methods suited for exploring population-scale microbiome datasets.
The research team, headed by Dr. ZHOU Haokui and supervised by Prof. ZHAO Guoping, at the Shenzhen Institutes of Advanced Technology (SIAT) of the Chinese Academy of Sciences developed tmap as an integrative framework for both pattern discovery and hypothesis generation for population-scale microbiome studies (Figure 1). Our method was currently published in Genome Biology, collected as an article in the special issue of Microbiome Biology.
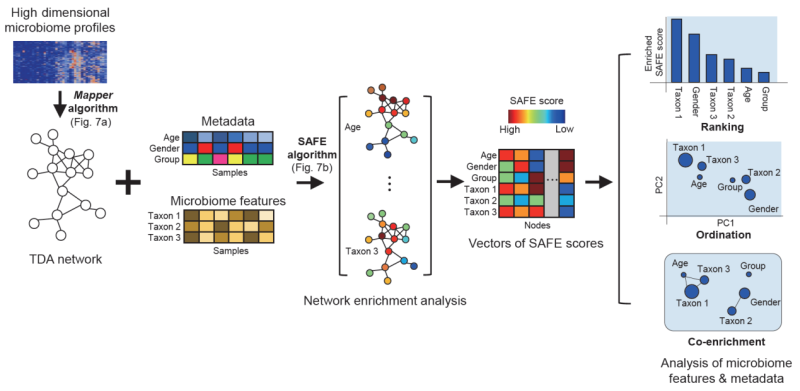
Figure 1. Overview of the tmap workflow for integrative microbiome data analysis. The workflow transforms high-dimensional microbiome profiles into a compressive topological network representation for microbiome stratification and association analysis.
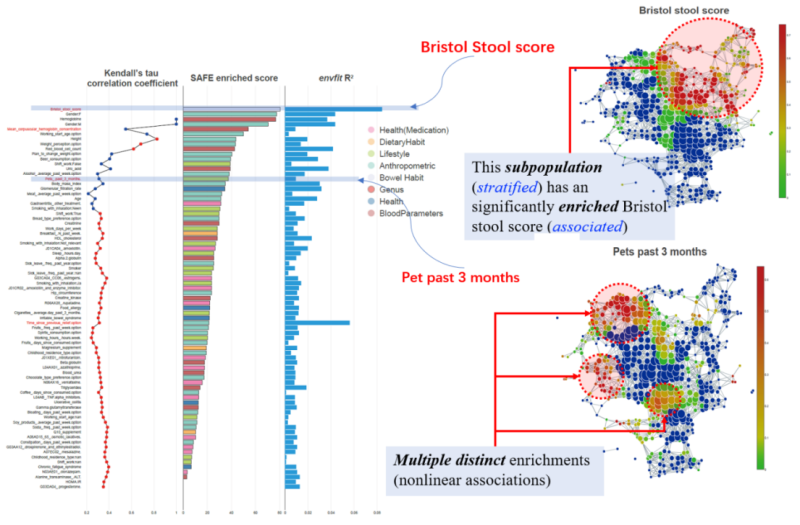
Figure 2. Stratification of the FGFP (Flemish Gut Flora project) microbiomes with host covariates. Linear patterns are observed for Bristol stool score by both envfit and tmap. However, nonlinear patterns of multiple local enrichments are observed for pets past 3 months, which is ranked differently between tmap and the conventional envfit method.
In tmap, we adopted the Mapper algorithm for topological data analysis, which allows us to construct an informative and compact network representation of high-dimensional microbiome dataset. With this advanced network-based data representation, tmap enables us to identify association of taxa or metadata within the network and to extract enrichment subnetworks of various association patterns. We applied tmap on both synthetic microbiomes and real-world microbiome datasets from the studies of human populations (such as the FGFP study, Figure 2) and earth environmental samples. We also compared tmap to conventional methods to demonstrate its superiority and effectiveness, which can be summarized as follows:
(1) tmap is able to integrate large-scale microbiome data with complex host phenotype metadata to systematically characterize the interrelations between host covariates and microbiome taxa, based on more efficient stratification and association analyses.
(2) tmap is capable in capturing both linear and nonlinear associations, which is superior to conventional methods. Based on a network representation of microbiome profiles, tmap is able to extract complex host-microbiome association patterns, providing insights to inform further hypothesis-driven studies.
tmap is provided as an open-source software at:
Detailed tutorials and online documents are available at: https://tmap.readthedocs.io/en/latest/
CONTACT:
ZHANG Xiaomin
Email: xm.zhang@siat.ac.cn
Tel: 86-755-86585299

