Exploring Functional Insights into the Human Gut Microbiome via the Structural Proteome
Nov 28, 2025
Despite rapid advances in microbiome research, a large proportion of proteins encoded by the human gut microbiome remain functionally uncharacterized. Sequence-based methods often fail to annotate these evolutionarily divergent proteins, leaving major gaps in our understanding of how microbial activities influence host metabolism, immunity, and disease. These "functional dark matter" highlights the need for frameworks that can explore protein function beyond sequence signals.
A research team led by Dr. DAI Lei at the Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, together with collaborators, published a research article in Cell Host & Microbe titled "Exploring Functional Insights into the Human Gut Microbiome via the Structural Proteome". This study established a structural proteome database and structure-based retrieval framework for the human gut microbiome, significantly improving the ability to predict functional "dark matter" such as phage proteins and host-derived bacterial isozymes.
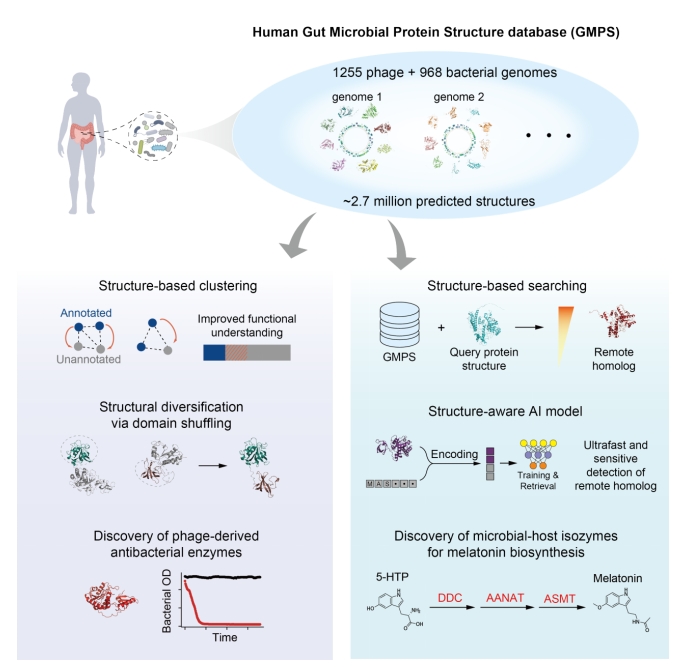
Using structure prediction tools, the researchers built the Human Gut Microbial Protein Structure Database (GMPS, https://www.gmpsdb.cn/), containing about 2.7 million predicted protein structures across 968 gut bacterial species and 1,255 phage genomes.
Researchers improved the annotation of phage proteins, of which up to 75% typically lack functional labels via sequence-based approaches. Structural analogy more than doubled the annotation rate and revealed extensive structural diversification of phage endolysins—antibacterial enzymes with high target-species specificity. Several newly predicted endolysins were experimentally validated and shown to eliminate gut pathobionts, demonstrating how structural proteomics can accelerate discovery of precision antimicrobials.
The study also highlights the value of structure-guided discovery of microbial–host isozymes—bacterial enzymes that perform host-like functions but are too sequence-divergent to detect using conventional tools. Through structural comparisons, the team identified previously unrecognized bacterial enzymes involved in melatonin biosynthesis. Biochemical assays validated their activities, and animal experiments showed that these microbial enzymes can modulate host melatonin levels and have direct impacts on host physiology.
To address cases where even structural similarity is insufficient, the researchers developed Dense Enzyme Retrieval (DEER), an alignment-free method powered by structure-aware language models. DEER enables ultrafast and sensitive detection of remote homologs, achieving state-of-the-art performance and extending functional annotation into regions previously inaccessible to both sequence and structure alignment-based tools.
Together, these advances establish a new paradigm that integrates large-scale structural genomics with artificial intelligence-driven inference to resolve the deep functional architecture of gut microbial communities. By illuminating substantial fractions of the microbiome's functional dark matter, this work opens new avenues for therapeutic discovery, precision microbiome engineering, and mechanistic understanding of microbe–host interactions.
Exploring Functional Insights into the Human Gut Microbiome via the Structural Proteome. (Image by SIAT)
File Download:
Media Contact
YU Rong
Email:
rong.yu@siat.ac.cn
rong.yu@siat.ac.cn
More on this News Release
JOURNAL
Cell Host & Microbe
DOI
10.1038/s41586-025-09509-7
Related Articles

May 23, 2022
The researchers longitudinally profiled the gut microbiota of mice to study the ecological basis for a baseline-dependent dynamic response to dietary fiber. From the time series data, they paramete...
Jan 13, 2022
The research group led by Prof. DAI Lei at the Shenzhen Institute of Advanced Technology (SIAT) of the Chinese Academy of Sciences, published their work “CRISPR/Cas-based genome editing for human ...